Chemical Kinetics 2
Shaun Williams, PhD
Reaction Mechanisms
- A reaction mechanism is a set of elementary steps that when taken together define a chemical pathway that connects the reactants to the products.
- An elementary reaction is one that proceeds by a single process
- molecular (or atomic) decomposition
- molecular collision
- The molecularity of an elementary reaction defines the order of the rate law for that reaction step.
Molecularity of Elementary Reactions
- Unimolecular elementary reactions have a single reactant \[ A\rightarrow \text{products} \]
- Bimolecular elementary reaction involve the collision of two reactants \[ A+B \rightarrow \text{products} \]
- Termolecular elementary reactions involve the collision of three reactants (these are rare for obvious reasons) \[ A+B+C \rightarrow \text{products} \]
- Another, more common form of termolecular elementary reactions involve two rapid steps \[ A+B \rightarrow AB^* \] \[ AB^* + C \rightarrow AB+C^* \]
The Requirements of a Reaction Mechanism
A valid reaction mechanism must satisfy three important criteria
- The sum of the steps must yield the overall stoichiometry of the reaction.
- The mechanism must be consistent with the observed kinetics for the overall reaction.
- The mechanism must account for the possibility of any observed side products formed in the reaction.
Example 12.1
For the reaction \[ A+B \rightarrow C \] is the following proposed mechanism valid? \[ A+A \xrightarrow{k_1} A_2 \] \[ A_2 + B \xrightarrow{k_2} C+A \]
Concentration Profiles for Some Simple Mechanisms
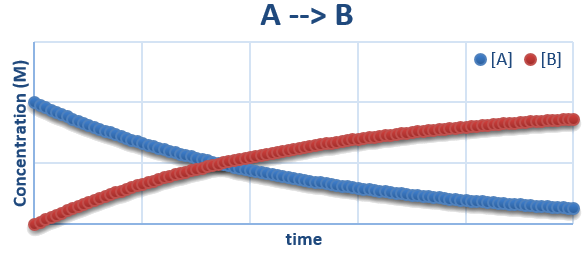
\(A \rightarrow B\)
- If this reaction is a single unimolecular elementary step then it can be writted as \[ A \xrightarrow{k_1} B \]
- Since it is an elementary step we can easily write the rate laws for it \[ \frac{d[A]}{dt} = -k_1[A] \] \[ \frac{d[B]}{dt} = +k_1[A] \]
A plot of \(A \rightarrow B\)
\( A \rightleftharpoons B \)
- The rate of change of the A and B will depend on both the forward and the backward reactions \[ A \xrightarrow{k_1} B \] \[ B \xrightarrow{k_{-1}} A \] or more typically \[ A \xrightleftharpoons{k_1}{k_{-1}} B \]
- The rate of change of concentration of A and B are \[ \frac{d[A]}{dt} = -k_1[A] + k_{-1}[B] \] \[ \frac{d[B]}{dt} = +k_1[A] - k_{-1}[B] \]
A plot of \(A \rightleftharpoons B\)
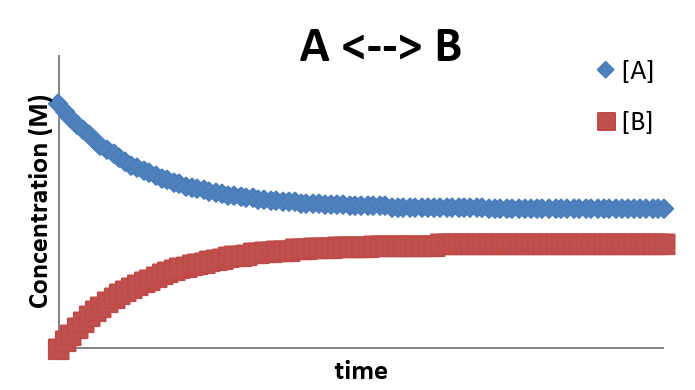
- This profile illustrates that even after the system achieves equilibrium, the forward and reverse reactions are still taking place.
- This is the nature of a dynamic equilibrium
At Equilibrium
- At equilibrium, the rate of formation of A and the rate of formation of B must be equal to zero \[ \frac{d[A]}{dt} = -k_1[A] + k_{-1}[B] = 0 \] \[ k_1[A] = k_{-1}[B] \] \[ \frac{k_1}{k_{-1}} = \frac{[B]}{[A]} \]
- So, the ratio of \(k_1\) to \(k_{-1}\) gives the value of the equilibrium constant, \(K_{eq}=\frac{[\text{products}]}{[\text{reactants}]} \)
\( A+C \rightarrow B+C \)
- Some reactions require a catalyst to mediate the reaction
- Catalysts are species that must be added (not formed as intermediates)
- Catalysts show up in the mechanism, usually in an early step
- Catalysts end up as part of the rate law
- Catalysts are reformed later so they don't appear in the overall reaction stoichiometry
- If the reaction \(A\rightarrow B\) is aided by a catalyst \(C\) then \[ A+C \rightarrow B+C \]
Kinetics of \(A+C \rightarrow B+C \)
- The rate of change of the concentrations is \[ \frac{d[A]}{dt} = -k[A][C] \] \[ \frac{d[B]}{dt} = +k[A][C] \] \[ \frac{d[C]}{dt} = -k[A][C] + k[A][C] = 0 \]
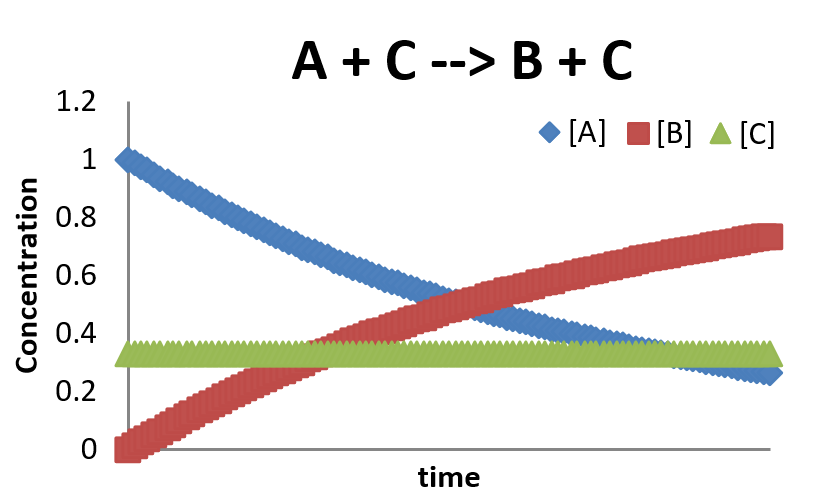
Final Anaylsis of \( A+C \rightarrow B+C \)
- This is a very simplified picture of reaction calaysis
- Generally, catalyzed reaction occur in at least two steps \[ A+B \rightarrow AC \] \[ AC \rightarrow B+C \]
- Later we will see how to handle this
\( A \rightarrow B \rightarrow C \)
- This mechanism features the formation of an intermediate
- Intermediates are species that are formed in one step and used in a later step
- Intermediates do not appeat in the reactions's stoichiometry
- Unlike catalysts, intermediates are not added to the reaction, but are created during the reaction.
- This reaction, \( A \xrightarrow{k_1} B \xrightarrow{k_2} C \) would have rate laws \[ \frac{d[A]}{dt} = -k_1[A] \] \[ \frac{d[B]}{dt} = k_1[A] - k_2[B] \] \[ \frac{d[C]}{dt} = k_2[B] \]
A plot of \(A \rightarrow B \rightarrow C\)

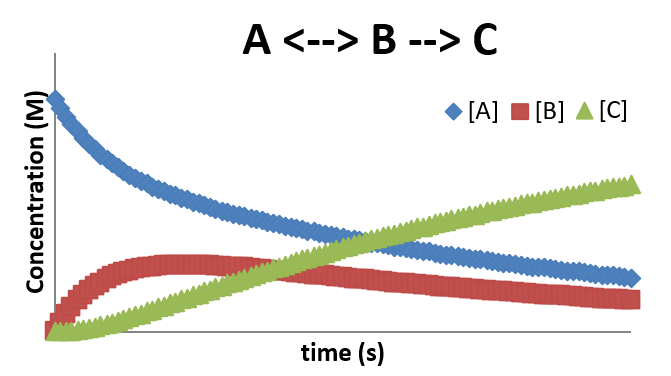
\(A \rightleftharpoons B \rightarrow C \)
- In many cases, the formation of an intermediate involves reversible step
- This step is sometimes called a pre-equilibrium step since it often will establish a near equilibrium while the reaction progresses
- This reaction can be broken down into two mechanistic steps \[ A \xrightleftharpoons{k_1}{k_{-1}} B \;\;\text{and}\;\; B \xrightarrow{k_2} C \] \[ \frac{d[A]}{dt} = -k_1[A]+k_{-1}[B] \] \[ \frac{d[B]}{dt} = k_1[A]-k_{-1}[B] -k_2[B] \] \[ \frac{d[C]}{dt} = k_2[B] \]
A plot of \(A \rightleftharpoons B \rightarrow C\)
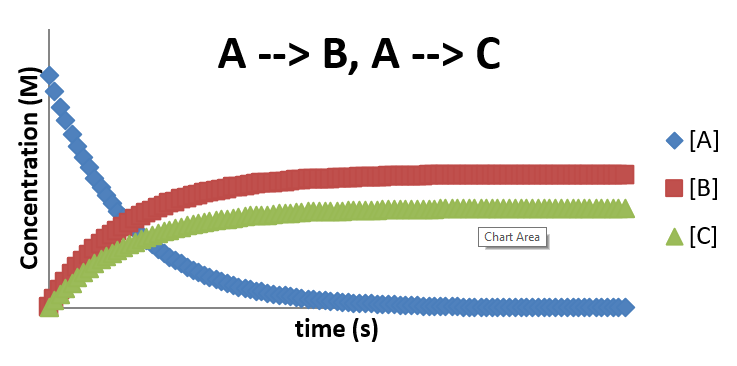
\(A \rightarrow B\) and \(A \rightarrow C\)
- There are many cases where a reactant can follow pathways to different products
- These pathways compete with each other
- A example is the following simple mechanism \[ A \xrightarrow{k_1} B \] \[ A \xrightarrow{k_2} C \] \[ \frac{d[A]}{dt} = -k_1[A]-k_2[A] \] \[ \frac{d[B]}{dt} = k_1[A] \] \[ \frac{d[C]}{dt} = k_2[A] \]
A plot of \(A \rightarrow B\) and \(A\rightarrow C\)
The Connection Between Reaction Mechanisms and Reaction Rate Laws
- Chemical kinetics gives us insights into the actual reaction pathways (mechanisms) that are followed
- Analyzing a reaction mechanism to determine the type of rate law that is consistant (or inconsistant) with a specific mechanism is important
An example: \(A+B\rightarrow C\)
- \[ \underbrace{A+A\xrightarrow{k_1}A_2}_{\text{step 1}} \] \[ \underbrace{A_2+B\xrightarrow{k_2} C}_{\text{step 2}} \]
- \[ \underbrace{A\xrightarrow{k_1}A^*}_{\text{step 1}} \] \[ \underbrace{A^*+B\xrightarrow{k_2} C}_{\text{step 2}} \]
Analysis of our example
- The first rate law will predict that the reaction should be second order in A.
- The second rate law will predict that the reaction should be first order in A.
- Based on the experimentally determined rate law's order, you can rule out one of these mechanisms.
- In order to analyze mechanisms and predict rate laws, we need some tools...
The Rate Determining Step Approximation
- The rate determining step approximation states that a mechanism can proceed no faster than its slowest step
- For example: \[ \underbrace{A+A\xrightarrow{k_1}A_2}_{\text{slow}} \;\;\text{and then} \;\; \underbrace{A_2+B\xrightarrow{k_2} C}_{\text{fast}} \]
- The rate determining step approximation says that the rate should be determined by the slow initial step \[ \frac{d[C]}{dt}=k_1[A]^2 \]
The Other Mechanism
- We can do the same for the other proposed mechanism \[ \underbrace{A\xrightarrow{k_1}A^*}_{\text{slow}} \] \[ \underbrace{A^*+B\xrightarrow{k_2} C}_{\text{fast}} \] so the rate law would be \[ \frac{d[C]}{dt} = k_1[A] \]
The Steady-State Approximation
- This is one of the most common approximations
- This approximation can be applied to the rate of change of concentration of a highly reactive (short lived) intermediate that holds a constant value over a long period of time
- The advantage is that for such an intermediate \(I\) \[ \frac{d[I]}{dt} = 0 \]
Our First Proposed Mechanism Again
- In our mechanism \[ A+A\xrightarrow{k_1}A_2 \] \[ A_2+B\xrightarrow{k_2} C \] \(A_2\) is an intermediate
- So, using the steady state approximation \[ \frac{d[A_2]}{dt} = k_1[A]^2-k_2[A_2][B] \approx 0 \]
- Thus \[ [A_2] \approx \frac{k_1[A]^2}{k_2[B]} \]
Further Simplification
\( [A_2] \approx \frac{k_1[A]^2}{k_2[B]} \)
- We can use that result to simplify our rate of product production \[ \begin{eqnarray} \frac{d[C]}{dt} &=& k_2[A_2][B] \\ &=& k_2 \left( \frac{k_1[A]^2}{k_2[B]} \right)[B] \\ &=& k_1[A]^2 \end{eqnarray} \]
Our Second Proposed Mechanism Again
- In our mechanism \[ A\xrightarrow{k_1}A^* \] \[ A^*+B\xrightarrow{k_2} C \] \(A^*\) is an intermediate
- So, using the steady state approximation \[ \frac{d[A^*]}{dt} = k_1[A]-k_2[A^*][B] \approx 0 \]
- Thus \[ [A^*] \approx \frac{k_1[A]}{k_2[B]} \]
Further Simplification to the Second
\( [A^*] \approx \frac{k_1[A]}{k_2[B]} \)
- We can use that result to simplify our rate of product production \[ \begin{eqnarray} \frac{d[C]}{dt} &=& k_2[A^*][B] \\ &=& k_2 \left( \frac{k_1[A]}{k_2[B]} \right)[B] \\ &=& k_1[A] \end{eqnarray} \]
Example 12.2
Use the steady-state approximation to derive the rate law for this reaction \[ \chem{2N_2O_5 \rightarrow 4NO_2 + O_2} \] assuming it follows the following three-step mechanism: \[ \chem{N_2O_5} \xrightleftharpoons{k_f}{k_b} \chem{NO_2 + NO_3} \] \[ \chem{NO_3 + NO_2} \xrightarrow{k_2} \chem{NO + NO_2 + O_2} \] \[ \chem{NO_3 + NO} \xrightarrow{k_3} \chem{2NO_2} \]
The Equilibrium Approximation
- As mentioned, in many cases, the formation of an intermediate involves a reversible step
- If the intermediate can reform reactants with a significant probability compared to forming products then we can utilize the equilibrium approximation
An Equilibrium Approximation Mechanism
- Consider the following mechanism \[ A+B \xrightleftharpoons{k_1}{k_{-1}} AB \] \[ AB \xrightarrow{k_2} C \]
- Due to the equilibrium, the steady state approximation would be combersome
- If the initial step is assumed to achieve equilibrium then \[ k_1[A][B]=k_{-1}[AB] \] \[ [AB]=\frac{k_1[A][B]}{k_{-1}} \]
Plugging in Our Approximation
\( [AB]=\frac{k_1[A][B]}{k_{-1}} \)
- We can now plug our approximation value into the rate of formation of our product \[ \begin{eqnarray} \frac{d[C]}{dt} &=& k_2[AB] \\ &=& k_2 \left( \frac{k_1[A][B]}{k_{-1}} \right) \\ &=& \frac{k_1k_2 [A][B]}{k_{-1}} \end{eqnarray} \]
Example 12.3
Given the following mechanism, apply the equilibrium approximation to the first step to predict the rate law suggested by the mechanism. \[ A+A \xrightleftharpoons{k_1}{k_{-1}} A_2 \] \[ A_2 + B \xrightarrow{k_2} C+A \]
Another Interesting Case
- Let's look at the following mechanism for the reaction \( A+2B\rightarrow 2C \) \[ A+B \xrightleftharpoons{k_1}{k_{-1}} I+C \] \[ I+B \xrightarrow{k_2} C \]
- Using the equilibrium approximation on \(I\) we find that \[ k_1[A][B] = k_{-1}[I][C] \;\;\Rightarrow\;\; [I]\approx \frac{k_1[A][B]}{k_{-1}[C]} \]
Simplifying Our New Case
\( [I]\approx \frac{k_1[A][B]}{k_{-1}[C]} \)
- We can substitute this into our expression for the rate of product formation \[ \begin{eqnarray} \frac{d[C]}{dt} &=& k_2[I][B] \\ &=& k_2 \left( \frac{k_1[A][B]}{k_{-1}[C]} \right) [B] \\ &=& \frac{k_1k_2[A][B]}{k_{-1}[C]} = k'\frac{[A][B]}{[C]} \end{eqnarray} \] where \( k'=\frac{k_1k_2}{k_{-1}} \)
- This rate law is first order in A and B but is negative 1 order in C.
The Lindemann Mechanism
- The Lindemann Mechanism is useful to demonstrate our kinetics techniques
- In the mechanism, a reactant is collisionally activated to an energetic state
- That energetic state can then return to reactants or form the products.
- Consider the following mechanism \[ A+A \xrightleftharpoons{k_1}{k_{-1}} A^* + A \] \[ A^* \xrightarrow{k_2} P \]
Applying the Steady State Approximation
- If we apply the steady state approximation to our intermediate \(A^*\) we find that \[ \frac{d[A^*]}{dt} = k_1[A]^2-k_{-1}[A^*][A]-k_2[A^*]\approx 0 \] \[ [A^*] \approx \frac{k_1[A]^2}{k_{-1}[A]+k_2} \]
- Substituting that into our rate of product formation \[ \begin{eqnarray} \frac{d[P]}{dt} &=& k_2[A^*] \\ &=& k_2 \left( \frac{k_1[A]^2}{k_{-1}[A]+k_2} \right) = \frac{k_1k_2[A]^2}{k_{-1}[A]+k_2} \end{eqnarray} \]
Analyzing the Result
\( \frac{d[P]}{dt}= \frac{k_1k_2[A]^2}{k_{-1}[A]+k_2} \)
- In the limit that \(k_{-1}[A]\ll k_2\) then \[ \frac{d[P]}{dt}= \frac{k_1k_2[A]^2}{k_2}=k_1[A]^2 \]
- In the limit that \(k_{-1}[A]\gg k_2\) then \[ \frac{d[P]}{dt}= \frac{k_1k_2[A]^2}{k_{-1}[A]}=\frac{k_1k_2[A]}{k_{-1}} \]
Third Body Collisions
- Sometimes a third-body collision is provided by an inert species \(M\) (such as argon in a gas reaction)
- In this case the mechanism could be \[ A+M \xrightleftharpoons{k_1}{k_{-1}} A^* + M \] \[ A^* \xrightarrow{k_2} P \]
- We can again use the stedy state approximation on \(A^*\) \[ \frac{d[A^*]}{dt} = k_1[A][M]-k_{-1}[A^*][M]-k_2[A^*]\approx 0 \] \[ [A^*] \approx \frac{k_1[A][M]}{k_{-1}[M]+k_2} \]
Rate of Product in Third Body Collisions
\( [A^*] \approx \frac{k_1[A][M]}{k_{-1}[M]+k_2} \)
- We can now insert this into our rate of product formation reaction \[ \begin{eqnarray} \frac{d[P]}{dt} &=& k_2[A^*] \\ &=& k_2 \frac{k_1[A][M]}{k_{-1}[M]+k_2} = \frac{k_1k_2[A][M]}{k_{-1}[M]+k_2} \end{eqnarray} \]
- If the concentration of \(M\) is constant then we can define a new effective rate constant, \(k_{uni}\) \[ k_{uni} = \frac{k_1k_2[M]}{k_{-1}[M]+k_2} \] \[ \frac{d[P]}{dt} = k_{uni}[A] \]
Analysis of the Effective Rate Constant
\( k_{uni} = \frac{k_1k_2[M]}{k_{-1}[M]+k_2} \)
- We can get very useful information about the individual steps we measure \(k_{uni}\) at various concentration of the third body collider \[ \frac{1}{k_{uni}} = \frac{k_{-1}[M]+k_2}{k_1k_2[M]} \] \[ \frac{1}{k_{uni}} = \frac{k_{-1}}{k_1k_2} + k_2\left(\frac{1}{[M]}\right) \]
- So, if we plot \(\frac{1}{k_{uni}}\) versus \(\frac{1}{[M]}\) we will get a straight line and
- The slope will be \(k_2\)
- The intercept will be \(\frac{k_{-1}}{k_1k_2}\)
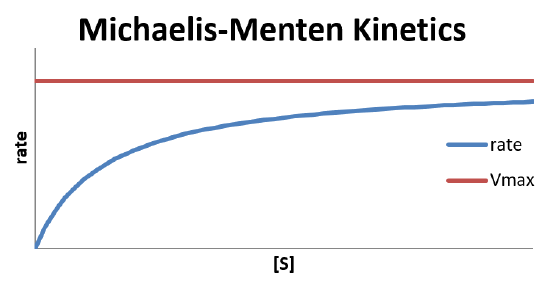
The Michaelis-Menten Mechanism
- The Michaelis-Menten mechanism is one which many enzyme mitigated reactions follow
- An enzyme (\(E\)) and a substrate (\(S\)) connect to form an enzyme-substrate complex (\(ES\))
- The enzyme-substrate complex degrades to form the product (\(P\))
- Overall, \(S\rightarrow P\) \[ E+S \xrightleftharpoons{k_1}{k_{-1}} ES \] \[ ES \xrightarrow{k_2} P+E \]
Analyzing Michaelis-Menten: The Beginning
- We first apply the equilibrium approximation to the first step \[ k_1[E][S] \approx k_{-1} [ES] \]
- Using mass conservation on the enzyme \[ [E]_0 = [E]+[ES] \;\;\Rightarrow\;\; [E]=[E]_0-[ES] \]
- Plugging this into our equilibrium approximation \[ k_1\left( [E]_0-[ES] \right)[S] = k_{-1} [ES] \] \[ k_1[E]_0[S]-k_1[ES][S] = k_{-1} [ES] \] \[ k_1[E]_0[S] = k_1[ES][S] + k_{-1} [ES] = \left(k_1[S]+k_{-1}\right)[ES] \] \[ \frac{k_1[E]_0[S]}{k_1[S]+k_{-1}}=[ES] \]
Analyzing Michaelis-Menten: Continuing
\( [ES]=\frac{k_1[E]_0[S]}{k_1[S]+k_{-1}} \)
- Plugging this into the rate of product formation \[\begin{eqnarray} \frac{d[P]}{dt} &=& k_2[ES] \\ &=& k_2 \left( \frac{k_1[E]_0[S]}{k_1[S]+k_{-1}} \right) \\ &=& \frac{k_1k_2[E]_0[S]}{k_1[S]+k_{-1}} \end{eqnarray}\]
- Multiply the top and bottom by \(\frac{1}{k_1}\) \[ \frac{d[P]}{dt} = \frac{k_2[E]_0[S]}{[S]+\frac{k_{-1}}{k_1}} \]
Analyzing Michaelis-Menten: Finishing
\( \frac{d[P]}{dt} = \frac{k_2[E]_0[S]}{[S]+\frac{k_{-1}}{k_1}} \)
- The ratio \(\frac{k_{-1}}{k_1}\) is the equilibrium constant that describes the dissociation of the enzyme-substrate complex, \(K_d\)
- The value \(k_2[E]_0\) is the maximum rate, \(V_{max}\)
- This means that we can write our equation as \[ \frac{d[P]}{dt} = \text{rate} = \frac{V_{max}[S]}{K_d+[S]} \]
Analyzing Michaelis-Menten with Steady-State Approximation: The Beginning
- Alternatively we can derive our expression using the steady-state approximation on \(ES\) \[ \frac{d[ES]}{dt} = k_1[E][S]-k_{-1}[ES] - k_2[ES]\approx 0 \] \[ [ES]=\frac{k_1[E][S]}{k_{-1}+k_2} = \frac{[E][S]}{K_m} \] where \(K_m=\frac{k_{-1}+k_2}{k_1}\) is the Michaelis constant.
- Plugging that is we find that \[ \frac{d[P]}{dt} = \frac{V_{max}[S]}{K_m+[S]} \]
Plot of Michaelis-Menten Kinetics
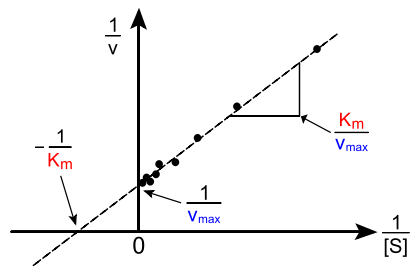
Lineweaver-Burk Plot
- Michaelis-Menten studies will measure the rate of the reaction as a function of substrate concentration
- We can rearrange our rate equation some that it becomes linear \[ \frac{1}{\text{rate}}=\frac{K_m+[S]}{V_{max}[S]} \] \[ \frac{1}{\text{rate}}=\frac{K_m}{V_{max}}\frac{1}{[S]}+\frac{1}{V_{max}} \]
A Lineweaver-Burk Plot Example
Chain Reactions
- A large number of reaction proceed through a series of steps that can collectively be classified as a chain reaction
- The reaction steps can be classified as
- initiation step - a step that creates the intermediates from stable species
- propagation step - a step that consumes an intermediate, but creates a new one
- termination step - a step that consumes intermediates without creating new ones
- These types of reations are very common when the interemediates involved are radiacals
Chain Reaction Example
- Consider the reaction \[ H_2+ Br_2 \rightarrow 2HBr \]
- The observed rate law for this reaction is \[ \text{rate} = \frac{k[H_2][Br_2]^\frac{3}{2}}{[Br_2]+k'[HBr]} \]
Chain Reaction Example Mechanism
- The proposed mechanism is \[ Br_2 \xrightleftharpoons{k_1}{k_{-1}} 2Br\cdot \] \[ 2Br\cdot + H_2 \xrightleftharpoons{k_2}{k_{-2}} HBr + H\cdot \] \[ H\cdot + Br_2 \xrightarrow{k_3} HBr + Br\cdot \]
Mechanism Analysis
- The change in concentrations of the intermediates can be written and the steady state approximation applied \[ \frac{d[H\cdot]}{dt} = k_2[Br\cdot][H_2]-k_{-2}[HBr][H\cdot]-k_3[H\cdot][Br_2]\approx 0 \] \[ \begin{align} \frac{d[Br\cdot]}{dt} = 2k_1[Br_2]&-2k_{-1}[Br\cdot]^2-k_2[Br\cdot][H_2] \\ &+k_{-2}[HBr][H\cdot]+k_3[H\cdot][Br_2] \approx 0 \end{align} \]
- Adding these two expression cancels the terms involving \(k_2\), \(k_{-2}\), and \(k_3\) \[ 2k_1[Br_2]-2k_{-1}[Br\cdot]^2=0 \]
Mechanism Analysis Continued
- Solving the equation from \(Br\cdot\) \[ [Br\cdot] = \sqrt{\frac{k_1[Br_2]}{k_{-1}}} \]
- This can be substituted into an expression for the \(H\cdot\) that is made by solving the steady state expression for \( \frac{d[H\cdot]}{dt}\) \[ [H\cdot] = \frac{k_2[Br\cdot][H_2]}{k_{-2}[HBr]+k_3[Br_2]} = \frac{k_2\sqrt{\frac{k_1[Br_2]}{k_{-1}}}[H_2]}{k_{-2}[HBr]+k_3[Br_2]} \]
Mechanism Analysis: Simplifying
- We can substitute are expression for \([H\cdot]\) and \([Br\cdot]\) into the rate of production of \(HBr\) \[ \begin{eqnarray} \frac{d[HBr]}{dt} &=& k_2[Br\cdot][H_2]+k_3[H\cdot][Br_2]-k_{-2}[H\cdot][HBr] \\ &=& k_2\sqrt{\frac{k_1[Br_2]}{k_{-1}}}[H_2]+k_3\left( \frac{k_2\sqrt{\frac{k_1[Br_2]}{k_{-1}}}[H_2]}{k_{-2}[HBr]+k_3[Br_2]} \right)[Br_2] \\ && -k_{-2}\left( \frac{k_2\sqrt{\frac{k_1[Br_2]}{k_{-1}}}[H_2]}{k_{-2}[HBr]+k_3[Br_2]} \right)[HBr] \end{eqnarray} \]
Mechanism Analysis: Simplifying This Time
- Let's try to actually simplify the expression \[ \text{rate} = k_2\sqrt{\frac{k_1[Br_2]}{k_{-1}}}[H_2] \left[ 1+\frac{k_3[Br_2]}{k_{-2}[HBr]+k_3[Br_2]}-\frac{k_{-2}[HBr]}{k_{-2}[HBr]+k_3[Br_2]} \right] \] \[ \text{rate} = k_2\sqrt{\frac{k_1[Br_2]}{k_{-1}}}[H_2] \left[ \begin{array}{l} \frac{k_{-2}[HBr]+k_3[Br_2]}{k_{-2}[HBr]+k_3[Br_2]} \\ +\frac{k_3[Br_2]}{k_{-2}[HBr]+k_3[Br_2]} \\ -\frac{k_{-2}[HBr]}{k_{-2}[HBr]+k_3[Br_2]} \end{array} \right] \]
Mechanism Analysis: Maybe this time!
- Now we can combine and start canceling some terms \[ \text{rate} = k_2\sqrt{\frac{k_1[Br_2]}{k_{-1}}}[H_2] \left[ \frac{k_{-2}[HBr]+k_3[Br_2]+k_3[Br_2]-k_{-2}[HBr]}{k_{-2}[HBr]+k_3[Br_2]} \right] \] \[ \text{rate} = k_2\sqrt{\frac{k_1[Br_2]}{k_{-1}}}[H_2] \left[ \frac{2k_3[Br_2]}{k_{-2}[HBr]+k_3[Br_2]} \right] \] \[ \text{rate} = \frac{2k_2k_3\sqrt{\frac{k_1}{k_{-1}}}[H_2][Br_2]^\frac{1}{2}}{k_{-2}[HBr]+k_3[Br_2]} \]
Mechanism Analysis: A Final Simplification
\( \text{rate} = \frac{2k_2k_3\sqrt{\frac{k_1}{k_{-1}}}[H_2][Br_2]^\frac{1}{2}}{k_{-2}[HBr]+k_3[Br_2]} \)
- Doing a little algebra we find that \[ \text{rate} = \frac{2k_2k_3\sqrt{\frac{k_1}{k_{-1}}}[H_2][Br_2]^\frac{1}{2}}{\frac{k_{-2}}{k_3}[HBr]+[Br_2]} = \frac{k[H_2][Br_2]^\frac{1}{2}}{k'[HBr]+[Br_2]} \] where \(k'=\frac{k_{-2}}{k_3}\) and \(k=2k_2k_3\sqrt{\frac{k_1}{k_{-1}}}\)
- This matches the experimental rate law
Catalysis
- There are many examples of reactions that involve catalysis
- One important example is from the environments, the catalytic decomposition of ozone \[ O_3 + O \rightarrow 2O_2 \]
- This reaction can be catalyzed by atomic chlorine
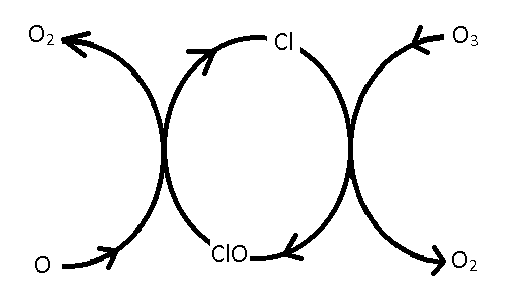
Mechanism of Ozone Decomposition
- The mechanism for the catalytic decomposition of ozone is \[ O_3 + Cl \xrightarrow{k_1} ClO+O_2 \] \[ ClO+O \xrightarrow{k_2} Cl + O_2 \]
- The rate of change of the intermediate (\(ClO\)) concentration is given by \[ \frac{d[ClO]}{dt} = k_1 [O_3][Cl] - k_2[ClO][O] \]
Analysis of Ozone Decomposition Mechanism
- Applying the steady state approximation to the intermediate \[ [ClO] = \frac{k_1[O_3][Cl]}{k_2[O]} \]
- The rate of production of the product, \(O_2\), is \[ \frac{d[O_2]}{dt} = k_2[O_3][Cl]+k_2[ClO][O] = k_2[O_3][Cl]+k_2\left( \frac{k_1[O_3][Cl]}{k_2[O]} \right)[O] \] \[ \frac{d[O_2]}{dt} = k_2[O_3][Cl]+ k_1[O_3][Cl] = \left(k_1+k_2\right)[O_3][Cl] \]
Schematic of Ozone Decomposition
Oscillating Reactions
- Most reaction convert reactant to products in a smooth process
- Some reactions show irregular behavior
- One particular phenomenon is that of oscillating reactions where the reactant concentrations rise and fall as the reaction progresses
- One way this happens is when the products of the reaction catalyze the reaction - this is called autocatalysis
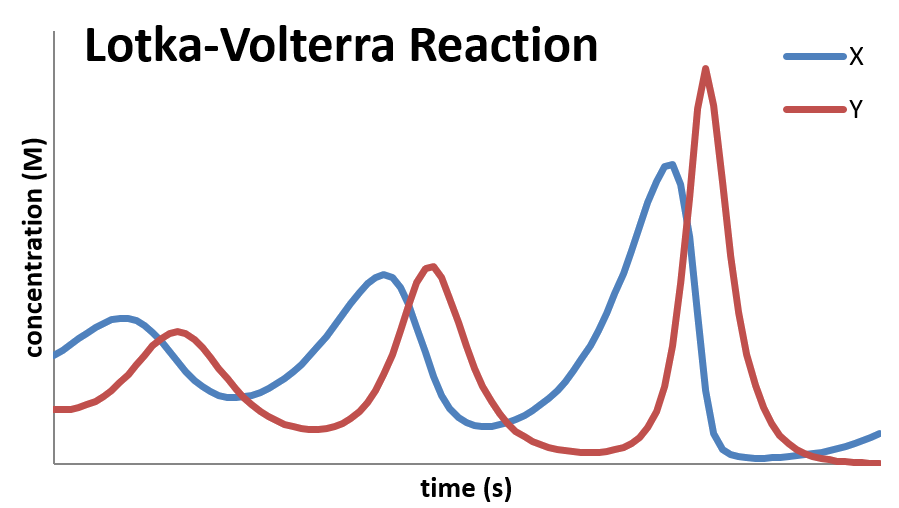
The Lotka-Voltera Mechanism
- An example of an autocatalyzed mechanism is the Lotka-Voltera mechanism \[ A+X \xrightarrow{k_1} X+X \] \[ X+Y \xrightarrow{k_2} Y+Y \] \[ Y \xrightarrow{k_3} B \]
- In this reaction, the concentration of A is held constant by continually adding it to the reaction mixture
- The first step is autocatalyzed so it speeds up in over time... as it the second step
Schematic of Lotka-Voltera Mechanism
/