Indroduction to Statistical Mechanics: Building Up to the Bulk
Shaun Williams, PhD
Atomic Structure
Atomic Structure
- A concept key to interpreting the second law of thermodynamics is the limitation by quantum mechanics on the number of states of our system
- The number of quantum states of any real system is a finite number
- This is an important difference between our classical understanding and our modern understanding
Quantization of Properties
- At molecular sizes, particles behave like waves
- To fulfill the mathematical rules of waves, parameters, such as energy and momentum, are only allowed certain quantized values
- For a moving particle particle, this quantization becomes measurable when the distance traveled is much greater than its de Broglie wavelength $$ \lambda_{dB} = \frac{h}{mv} $$
- In this equation, called the de Broglie wavelength equation
- \(h\) is Planck’s constant, \( 6.626 \times 10^{-34}\,\mathrm{J}\cdot \mathrm{s}\)
- \(m\) is the particles mass
- \(v\) is the particles velocity
Quantizer Energy
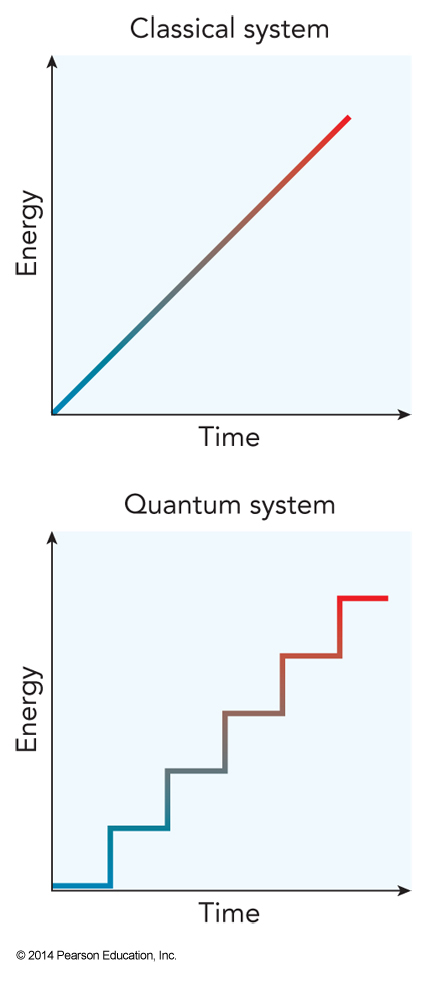
- In a classical system, energy is a continuous function
- In a quantum system, energy comes only in discrete values
Problem with the quantum nature
- When dealing with waves, the particles/waves cannot have their position pinned down to a single location
- For any parameter, \(A\left(\vec{r}\right)\), the average value of this parameter is given by the average value theorem
$$ \expect{A} = \int_{\text{all space}} \mathcal{P}_\vec{r}\left(\vec{r}\right) A\left(\vec{r}\right) d\vec{r} $$
- Where \(\mathcal{P}_\vec{r}\left(\vec{r}\right)\) is the probability distribution
Particle in a Three-Dimensional Box
- Consider a 3-D box with lengths of \(a\), \(b\), and \(c\) along the \(x\), \(y\), and \(z\) axes, respectively
- Possible energy values for a particle in the box is $$ \begin{align} \varepsilon_{n_x,n_y,n_z} &= \frac{\pi^2 \hbar^2}{2m}\left( \frac{n_x^2}{a^2}+\frac{n_y^2}{b^2}+\frac{n_z^2}{c^2} \right) \\ &\approx \frac{\pi^2\hbar^2}{2m(abc)^\bfrac{2}{3}}\left( n_x^2+n_y^2+n_z^2 \right) = \varepsilon_0n^2 \end{align} $$
Energy Levels of a Three-Dimensional Box
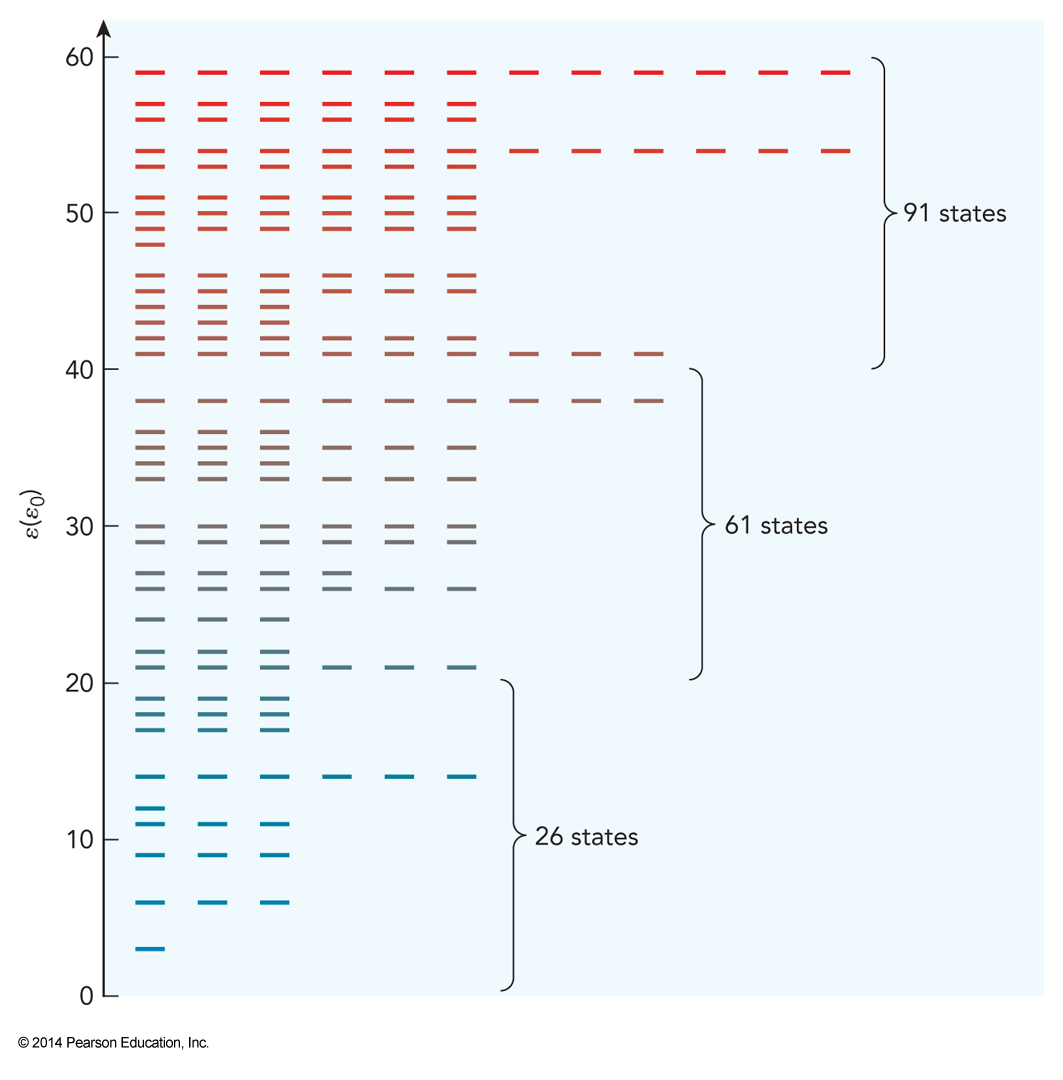
States of Such a Particle
- A particle which has the lowest values of \(n_x\), \(n_y\) and \(n_z\) is said to be in its ground state
- Any other state is said to an excited state
- When two or more quantum states share the same energy, we say they are degenerate
- (2,1,1), (1,2,1), and (1,1,2)
- Correspondence principle – quantum mechanics approaches classical mechanics as we increase the particle’s kinetic energy, mass, or domain
Use of Quantum Mechanics
- Due to correspondence principle:
- Use QM when \(\lambda_{dB}\) is comparable to the domain size
- Use classical when \(\lambda_{dB}\) is much smaller
- Entropy allows us to indirectly measure the density of quantum states, \(W(\varepsilon)\)
- The density of states tells how many quantum states near energy \(\varepsilon\) there are per unit energy
Quantum States of Atoms
- The same analysis can be used on one-electron atoms yielding $$ E_n = -\frac{Z^2m_ee^4}{2\left(4\pi \epsilon_0\right)^2n^2\hbar^2} \equiv -\frac{Z^2}{2n^2}E_h $$
- \(E_h\) is the energy unit Hartree
- Hartrees are convenient units for calculations involving electrons in atoms
- Conversion: \(1\,E_h=4.360\times 10^{-18}\,\mathrm{J}\)
Energy Levels of the Hydrogen Atom
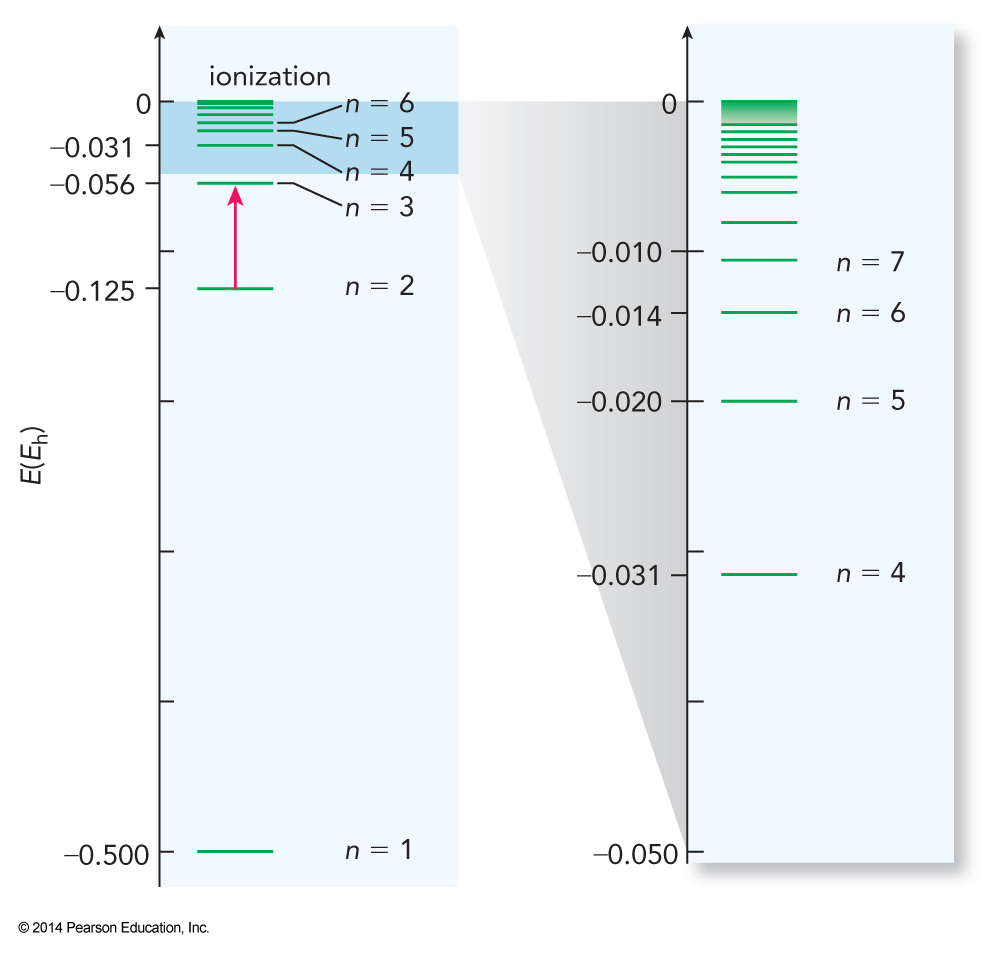
Quantum State of Electrons
- The quantum state of electrons depend on three other quantum numbers
- \(l\) – angular momentum quantum number; defines the shape
- \(m_l\) – magnetic quantum number; defines orientation in space
- \(m_s\) – spin angular momentum
- For each atom, we can specify the entire set of quantum numbers (electron configuration)
Term Symbols
- In addition to electron configurations, which give \(n\) and \(l\) values, we can specify the term symbol which gives \(m_l\) and \(m_s\) values
- Ground state of nitrogen is \( 1s^22s^22p^3\) and it has a term symbol of \({}^4S\)
Electron Spin
- Spin is really an intrinsic property, like mass and charge
- Particles called Fermions all have spins that are half-integers
- Electrons, protons, neutrons
- Particles called Bosons all have integer spins
- \(\chem{{}^4He}\) nucleus
The Pauli Exclusion Principle
- No two individual Fermions can have the same set of quantum numbers
- Unlike Fermions, according to the Pauli exclusion principle, multiple Bosons can have the same set of quantum numbers
- This gives rise to very interesting effects (chapter 4)
Degrees of Freedom
- Atoms in molecules attract and repel.
- Energy added to a molecule goes into one of 4 forms of motion
- Electronic – fast motion
- Vibrational
- Rotational
- Translational – slow motion
- Note: increasing mass of the moving particle
Translational Energy
- Due to its large mass and slow speed, translational energy can normally be treated classically $$ K=\frac{mv^2}{2} $$
- We don’t need to worry too much about the other forms yet
- When energy is added to a system, it distributes itself into some or all of these degrees of freedom
Electronic State Transitions (UV-vis)
- Quantum states of molecules is not so easy
- Exciting from the electronic ground state to excited state gives us information
- This is done with electromagnetic (EM) radiation
- A photon has energy given by Planck’s law
$$ E_{photon} = h\nu = \frac{hc}{\lambda} $$
- \(c\) – speed of light: \(c=2.9979\times 10^8\,\mathrm{m}\,\mathrm{s}^{-1}\)
Vibrational State Transitions (IR)
- After electronic, the remaining contributions come from the motion of the nuclei
- For \(N_{atom}\) in the molecule, there are a total of \(3N_{atom}\) degrees of freedom
- Number of vibrational degrees of freedom:
- For non-linear molecules: \(3N_{atom}-6\)
- For linear molecules: \(3N_{atom}-5\)
Vibrational Modes
- For each vibrational coordinate we can define a vibrational constant, \(\omega_e\)
- Depends on mass of atoms and rigidity of bond
- Harmonic approximation defined the constant
$$ \omega_e(J)=\hbar \sqrt{\frac{k}{\mu}} $$
- The reduced mass is \(\mu=\frac{m_Am_B}{m_A+m_B}\)
Vibrational Energies
- The quantum states for vibrational motion have vibrational energies approximately equal to
$$ E_v=\left(v+\frac{1}{2}\right)\omega_e $$
- \(v\) can be any integer 0 or greater
- Vibrational energies correspond to the infrared region of the EM spectrum
Rotational States Transions (microwave)
- Non-linear molecules have three rotational coordinates
- Linear molecules have two rotational coordinates
- For a simple linear molecule the rotational energies are given by
$$ E_J=BJ(J+1) $$
- \(B\) is the rotational constant
- \(J\) can be any integer 0 or greater
Rotational State Degeneracies
- Rotational states have degeneracies corresponding to different orientations of rotational motion
- For linear molecules, \(g_{rot}=2J+1\)
- We will eventually need to know the degeneracy to determine how a molecule distributes energy
Bulk Properties
Some Definitions
- System (or sample) – the thing we want to study
- Surroundings – everything outside the system
- Boundary – what separates the system from the surroundings
- Universe – everything: the system, the surroundings, and the boundary between them
Balloon Example
- If we discuss the properties of the gas in a balloon
- The gas in the balloon is the system
- The atmosphere around the balloon is the surroundings
- The elastic material of the walls of the balloon is the boundary
- The boundary defines how the system interacts with the surroundings
Bulk
- In bench-top chemistry, we deal with so many particles that removal of 30 or addition of 10000 will not change the properties
- A cluster of 8 sodium atoms has an ionization energy 20% higher than the bulk metal value
- This drops to 10% if we use 9 sodium atoms
- Bulk limit is much smaller than a visible sample
- \(\sim 10^{10}\,\mathrm{molecules} < 10^{-13}\,\mathrm{moles}\)
Difficulty with QM
- QM accurately predicts the properties of individual molecules
- QM will work as the system becomes bigger
- It is no longer practical to analyze a mole of particles with QM, simply to complicated
- With so many molecules, we typically don’t care how a single one of them behaves
Macroscopic Properties
- We seek macroscopic properties of the system
- Two group of macroscopic properties:
- Extensive – obtained by summing together contributions from all the molecules in the system
- Intensive – obtained by averaging the contributions from all the molecules in the system
Parameters
- Extensive Parameters
- \(N\), the total number of molecules in the system or the total number of moles, \(n\)
- \(V\), the total space occupied
- \(E\), the sum of the translational, rotational, vibrational, and electron energies
- Intensive Parameters
- \(P\), the pressure, which is the average force per unit area exerted by the molecules on their surroundings
- \(\rho\), the number density, which is the average number of molecules per unit volume
- Related to mass density $$ \rho_m=\rho\frac{\mathcal{M}}{\mathcal{N}_A} $$
Statistical Mechanics
- Statistical mechanics used probability theory to describe the macroscopic groups of molecules
- Based on common characteristics of individual molecules
- Statistical mechanics extends the results of QM to the behavior of bulk quantities of substances
- The things we want to study are perfect statistical samples
Common Assumptions in Statistical Mechanics
- Chemically identical molecules share the same physics
- Macroscopic variables are continuous variables
- Measured properties reflect the ensemble average
- If we fix some values (e.g. volume and moles)
- Ensemble - Set of all quantum states, same values
- Microstate – a unique quantum state
- \(\Omega\) – number of microstates in the ensemble
Ergodic Hypothesis
- If we fix \(E\), \(N\), and \(V\)
- Let the system find its own pressure
- The result will be the average of \(P\) over all microstates
- The ensemble average
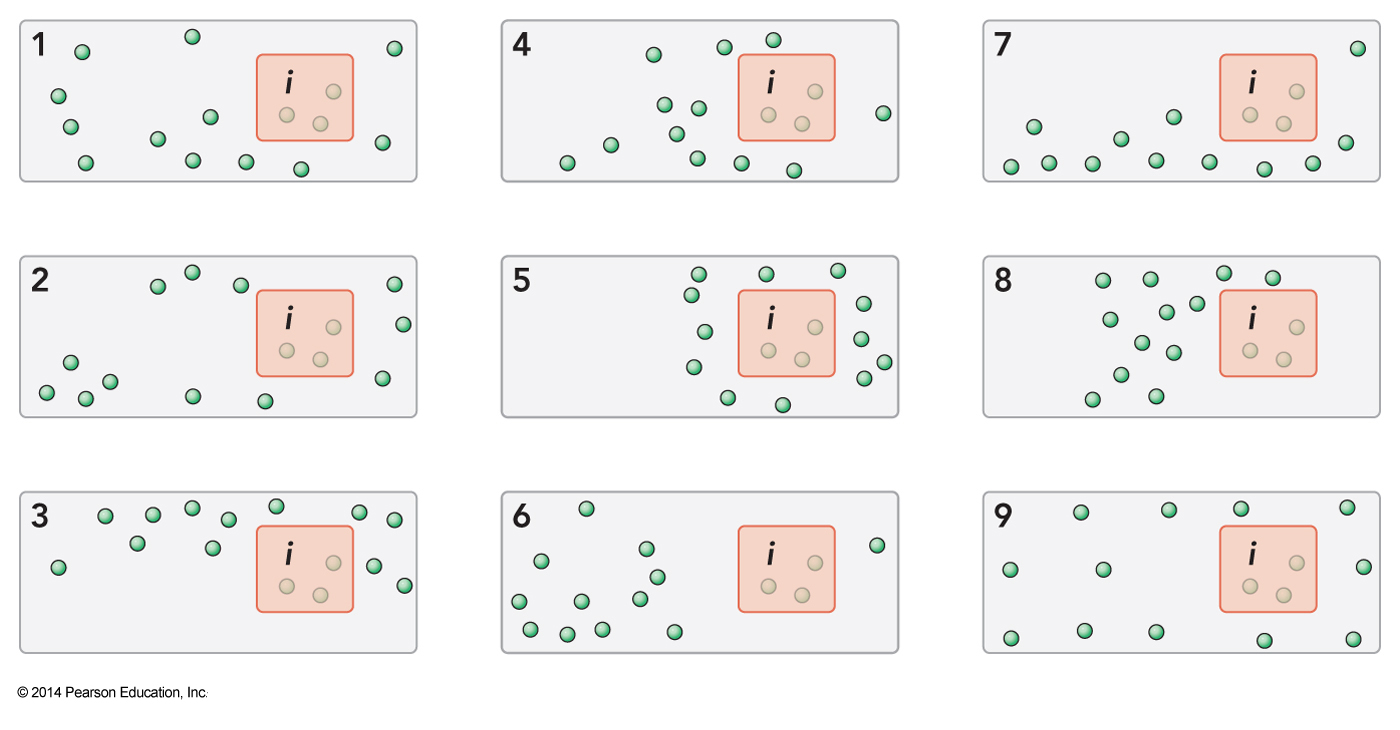
12-Member Ensemble
- Here are 12 distinct microstates
- All have the same properties
- Let the particle exert a pressure of p0 on the walls next to it
- Half the top walls have two particles next to it and half have one
- \(\expect{p_{top}} = 1.5p_0\)
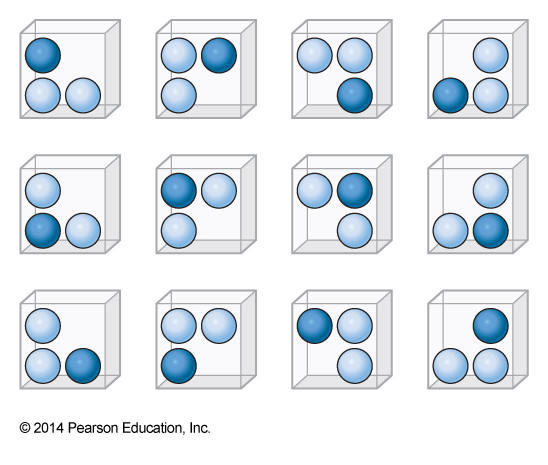
The Microcanonical Ensemble
- Approaching a problem, we must decide which parameters are allowed to vary
- More flexible is more realistic but more difficult to solve
- If we fix all the extensive variables, \(E\), \(V\), and \(N\) we have our first ensemble – the microcanonical ensemble
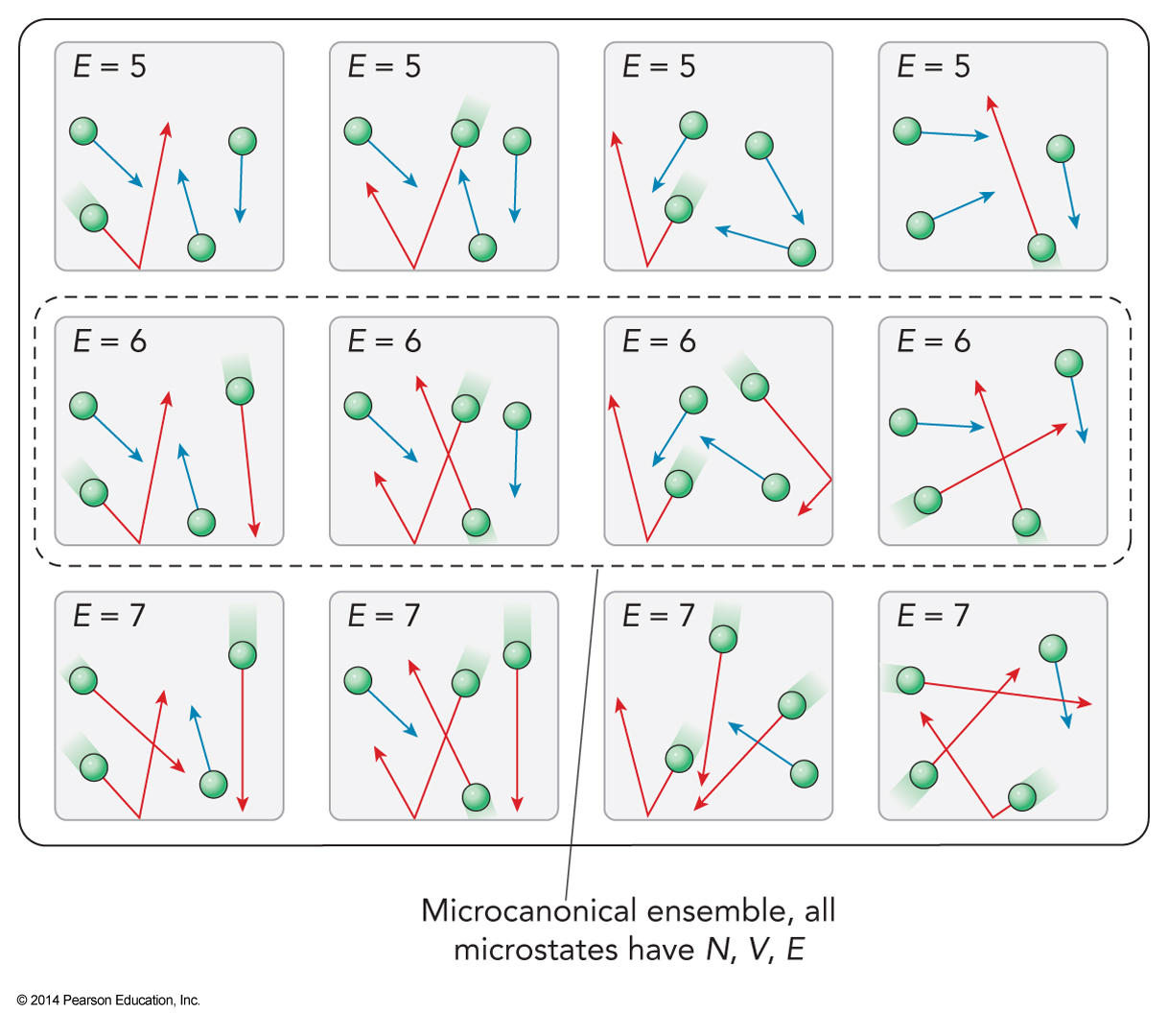
Properties of Microcanonical Ensemble
- If we fix all extensive variables then intensive variables that are ratios of extensive variables must also be fixed
- Density, \(\rho=\frac{N}{V}\)
- What can change?
- Microscopic properties that we’re not bothering to measure
- Some intensive properties (such as pressure) that are not ratios of extensive variables
Advantages of Microcanonical Ensemble
- Energy and mass are rigorously conserved
- There system must be completely isolated from the rest of the universe
- Constant volume means that no mechanical force pushes on the surroundings
- Isolation greatly simplifies the system
Entropy
Boltzmann Entropy
- The Boltzmann entropy is given by
$$ S\equiv k_\mathrm{B}\ln \Omega $$
- Boltzmann constant is \(k_\mathrm{B}=1.381\times 10^{-23}\,\mathrm{J}\,\mathrm{K}^{-1}\)
- This provides a different definition of the relation between heat and temperature
- This will be our rigorous definition of entropy since it works under and circumstances
- The entropy counts the total number of distinct microstates of the system
Entropy and Microstates
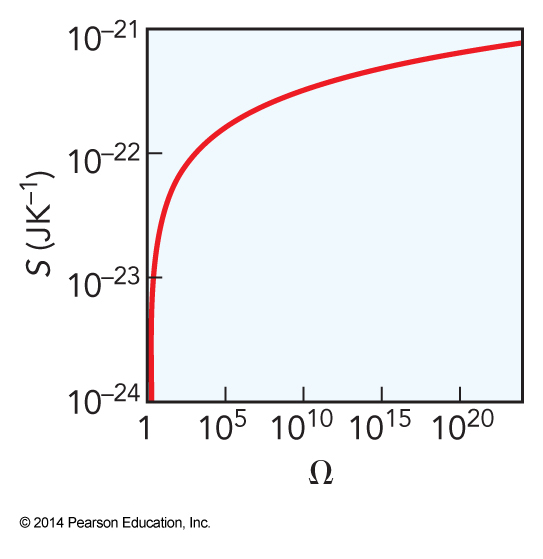
\(\Omega\) versus \(g\)
- For a microcanonical ensemble
- \(\Omega\) is the total number of microstates that have the same energy
- Under these conditions \(\Omega\) is the same as \(g\)
- \(\Omega\) is the total number of microstates in an ensemble
- \(g\) is the degeneracy of quantum states in some particular individual particle
Size of \(\Omega\)
- \(\Omega\) is enormous
- However, \(\Omega\) is always finite
- The number of arrangements of system is countable
- In a classical system, \(\Omega\) would be infinite due to the continuous nature of classical mechanics
- Quantization makes \(\Omega\) finite
Two Non-Interacting Subsystems
- The degeneracy is the product of degeneracies
- The entropy is the sum of the entropies $$ \begin{align} \Omega_{\chem{A+B}} &= \Omega_\chem{A}\Omega_\chem{B} \\ \ln \Omega_{\chem{A+B}} &= \ln \left(\Omega_\chem{A}\Omega_\chem{B}\right) = \ln \Omega_\chem{A}+\ln \Omega_\chem{B} \\ S_\chem{A+B} &= S_\chem{A}+S_\chem{B} \end{align} $$
Gibbs Definition of Entropy
- Gibbs developed a general equation for estimating the entropy in any ensemble using probability rather than \(\Omega\)
- Derivation:
- The total number of ways of arranging the labels on \(N\) molecules is \(N!\)
- There are a total of \(N_i!\) ways of rearranging the labels
Example of Calculating \(\Omega\)
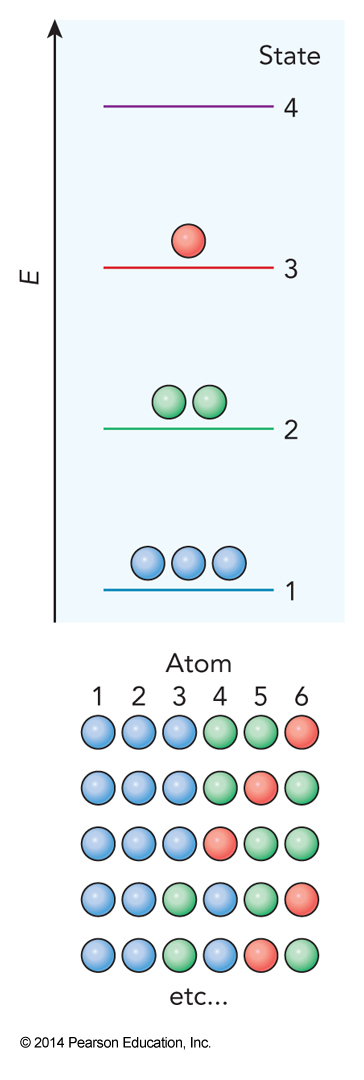
The Ensemble Size
- These definitions mean that we get $$ \Omega=\frac{N!}{N_1!N_2!N_3!\dots N_k!} = \frac{N!}{\prod_{i=1}^kN_i!} $$
- Example: 6 particles with 4 possible states with 3 in state 1, 2 in state 2, 1 in state 3 and 0 in state 4 $$ \Omega = \frac{6!}{(3!)(2!)(1!)(0!)}=60 $$
- Using this definition of the size of the ensemble we find $$ \begin{align} S &= k_\mathrm{B} \ln \Omega = k_\mathrm{B} \ln \left( \frac{N!}{\prod_{i=1}^kN_i!} \right) \\ &= k_\mathrm{B} \left[ \ln N! - \ln \prod_{i=1}^k N_i! \right] \end{align} $$
Using Stirling's Approximation
$$ S = k_\mathrm{B} \left[ \ln N! - \ln \prod_{i=1}^k N_i! \right] $$
- Now we can use Stirling’s approximation: \( \ln N! \approx N\ln N\) to yield $$ \begin{align} S &= k_\mathrm{B} \left[ N\ln N - N - \left( \sum_{i=1}^k N_i \ln N_i - \sum_{i=1}^k N_i \right) \right] \\ &= k_\mathrm{B} \left[ \sum_{i=1}^k N_i \ln N - N - \sum_{i=1}^k N_i \ln N_i +N \right] \\ &= k_\mathrm{B} \sum_{i=1}^k \left[ N_i \ln N - N_i \ln N_i \right] \\ &= k_\mathrm{B} \sum_{i=1}^k N_i \left[ \ln N - \ln N_i \right] \end{align} $$
Rewriting Entropy in Terms of Probability
$$ S = k_\mathrm{B} \sum_{i=1}^k N_i \left[ \ln N - \ln N_i \right] $$
- We can write this in terms of probabilities \(\mathcal{P}(i)=\frac{N_i}{N}\) $$ \begin{align} S &= Nk_\mathrm{B} \sum_{i=1}^k \frac{N_i}{N} \ln \frac{N}{N_i} \\ &= Nk_\mathrm{B} \sum_{i=1}^k \mathcal{P}(i) \ln \frac{1}{\mathcal{P}(i)} \\ &= -Nk_\mathrm{B} \sum_{i=1}^k \mathcal{P}(i) \ln \mathcal{P}(i) \end{align} $$
Temperature and the Partition Function
The Ideal Gas
- A collection of particles
- Don’t interact with each other at all
- Elastically bounce off walls
- We want to use statistical mechanics to find the pressure exerted by an ideal gas on the walls of the container as a function of the macroscopic variables
System and Reservoir
- The box has:
- Fixed volume, \(V\)
- Fixed number of molecules, \(N\)
- Energy will no longer be held constant
- Reservoir strongly interacts with system
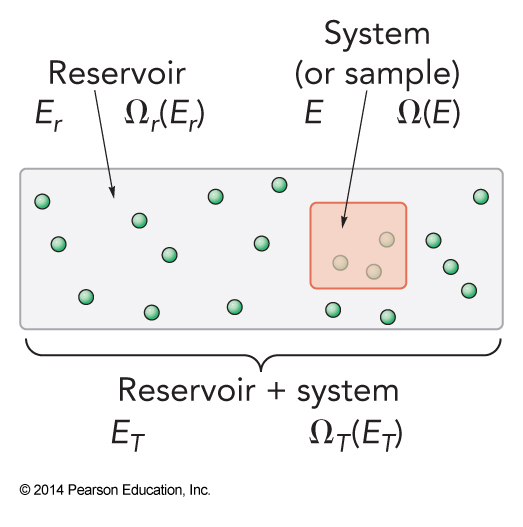
The Reservoir
- The reservoir has:
- \(E_r \gg E\)
- \(E+E_r = E_T \gg E\)
- \(\Omega_r\left(E_r\right) \gg \Omega\left(E\right)\)
- The ensemble size of the universe is $$ \Omega_T\left(E_T\right) = \sum_E \Omega\left(E\right) \Omega_r \left(E_r\right) $$
\(\mathcal{P}(i)\) and Number of Microstates
- Choose a single microstate, \(i\), with energy \(E_i\)
- If there are 9 distinct microstates with energy the reservoir must be changing
- Thus we can determine the system number from the reservoir number (\(E_r=E_T-E\))
- The probability of the system being in state \(i\) is $$ \mathcal{P}(i)=\frac{\Omega_r\left(E_r\right)}{\Omega_T\left(E_T\right)} $$
Ensemble Average
A Problem
- The problem is we don’t want to have to measure the reservoir instead of the system
- First we will rewrite this in terms of entropy (Taylor series expansion) $$ S_r \left( E_r \right) = S_r \left( E_T-E_i \right) = S_r \left( E_T \right) - \left. \left( \frac{\partial S_r}{\partial E_r} \right)_{V,N} \right|_{E_T} E_i + \cdots $$
- For a fixed total energy we know that \(dE_T = d\left(E_T-E\right)=-dE\)
- We also know that \(\Omega_T\left(E_T\right)=\Omega_r\left(E_r\right)\Omega\left(E\right)\)
Continuing to Solve the Problem
- We also know that $$ \begin{align} dS_r &= k_\mathrm{B} d \ln \Omega_r \left(E_r\right) = k_\mathrm{B} d\left[ \ln \left( \frac{\Omega_T\left(E_T\right)}{\Omega\left(E\right)} \right) \right] \\ &= k_\mathrm{B} d \left[ \ln \Omega_T \left(E_T\right) - \ln \Omega\left(E\right) \right] = -k_\mathrm{B} d\ln \Omega\left(E\right) \\ &= -dS \end{align} $$
- Combining these equations we find that $$ \left. \left( \frac{\partial S_r\left(E_r\right)}{\partial E_r} \right) \right|_{E_T} = \left. \left( \frac{\partial S(E)}{\partial E} \right) \right|_{E_T} $$
Rewriting the Entropy Equation
- Using these previous equation we find $$ S_r\left(E_r\right) = S_r\left(E_T-E_i\right) = S_r\left(E_T\right) - \left. \left( \frac{\partial S(E)}{\partial E} \right) \right|_{E_T} E_i $$
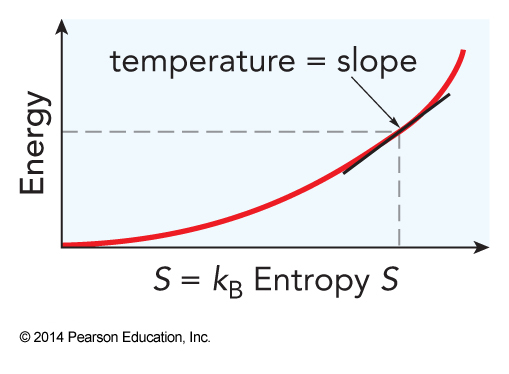
- This introduces temperature $$ T \equiv \left( \frac{\partial E}{\partial S} \right)_{V,N} $$
- Using temperature in the entropy equation $$ S_r\left(E_r\right) = S_r\left(E_T-E_i\right)=S_r\left(E_T\right)-\frac{E_i}{T} $$
Solving the Issues
- We can now go back to \(\Omega\) $$ k_\mathrm{B} \ln \left[ \Omega_r\left(E_r\right) \right] = k_\mathrm{B} \ln \left[ \Omega_r\left(E_T\right) \right] -\frac{E_i}{T} $$
- Solving for the number of reservoir states we get $$ \Omega_r\left(E_r\right)=\Omega_r\left(E_T\right)e^{-\bfrac{E_i}{k_\mathrm{B}T}} $$
Temperature
A New Ensemble
- In typical experiments, the energy is not fixed
- The system and the surroundings/reservoir can exchange energy
- If the reservoir is big enough, its properties are constant, so we fix \(T\)
- So a canonical ensemble has \(T\), \(V\), and \(N\) fixed and \(E\) as a variable
More Equations, Now Within the Canonical Ensemble
- Combining some previous equations $$ \mathcal{P}(i)=\frac{\Omega_r\left(E_T\right)}{\Omega_T\left(E_T\right)} e^{-\bfrac{E_i}{k_\mathrm{B}T}} $$
- The \(\Omega\) are constants
- We can eliminate them by requiring that the probability be normalized $$ \sum_{i=1}^\infty \mathrm{P}(i) = \frac{\Omega_r\left(E_T\right)}{\Omega_T\left(E_T\right)} \sum_{i=1}^\infty e^{-\bfrac{E_i}{k_\mathrm{B}T}} = 1 $$
Solving the Equation
$$ \sum_{i=1}^\infty \mathrm{P}(i) = \frac{\Omega_r\left(E_T\right)}{\Omega_T\left(E_T\right)} \sum_{i=1}^\infty e^{-\bfrac{E_i}{k_\mathrm{B}T}} = 1 $$
- Solving for our fraction $$ \frac{\Omega_r\left(E_T\right)}{\Omega_T\left(E_T\right)} = \frac{1}{\sum_{i=1}^\infty e^{-\bfrac{E_i}{k_\mathrm{B}T}}} = \frac{1}{Q(T)} $$
- We have made a definition of the partition function
Canonical Partition Function and Distribution
- So, the canonical partition function is $$ Q(T)=\sum_{i=1}^\infty e^{-\bfrac{E_i}{k_\mathrm{B}T}} $$
- The canonical distribution can be written as $$ \mathcal{P}(i) = \frac{e^{-\bfrac{E_i}{k_\mathrm{B}T}}}{Q(T)} $$
Meaning
$$ \mathcal{P}(i) = \frac{e^{-\bfrac{E_i}{k_\mathrm{B}T}}}{Q(T)} $$
- We see the importance of \(T\)
- The likelihood of getting the system into a particular state \(i\) drops exponentially with respect to the ratio of the microstate energy \(E_i\) to the thermal energy \(k_\mathrm{B}T\)
- The temperature establishes how likely the system is to be found at any particular energy.
Distribution in a 3D Box
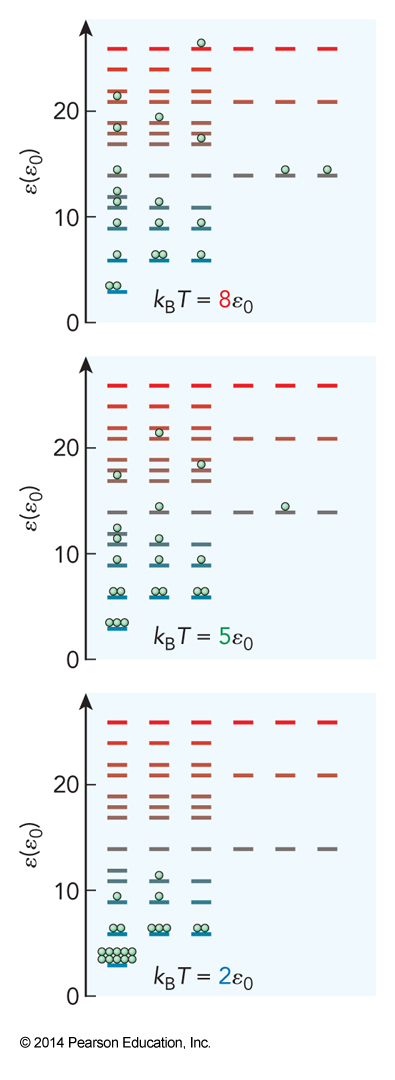
Finding Another Likelihood
- Let’s find the likelihood of finding the system t any particular energy E $$ \mathcal{P}(E)=\Omega(E)\mathcal{P}(i) = \frac{\Omega(E) e^{-\bfrac{E_i}{k_\mathrm{B}T}}}{Q(T)} $$
- In all these equation the beauty of the canonical ensemble is that is allows us to ignore how \(\Omega\) depends on \(V\) and \(N\)
Assertion
- The probability extends to the probability of a particle within our system having some particular energy \(\varepsilon\)
- Say we have a particular quantum state \(i\) and degeneracy \(g\) $$ \begin{align} \mathcal{P}(\varepsilon) &= \frac{g(E) e^{-\bfrac{\varepsilon}{k_\mathrm{B}T}}}{q(T)} \\ q(T) &= \sum_{\varepsilon=0}^\infty g(E) e^{-\bfrac{\varepsilon}{k_\mathrm{B}T}} \end{align} $$
Rotational Partition Functions
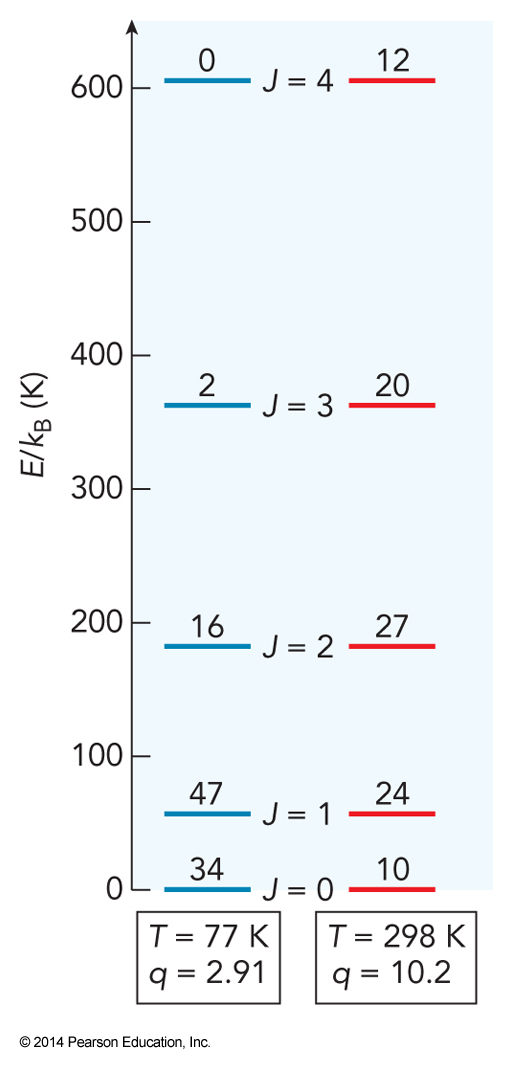
Check of Zero Energy
- If we measure all energy relative to \(E_0\) then $$ \begin{align} \mathcal{P}(E) &= \frac{ge^{-\bfrac{\left(E-E_0\right)}{k_\mathrm{B}T}}}{\sum_E ge^{-\bfrac{\left(E-E_0\right)}{k_\mathrm{B}T}}} \\ &= \left( \frac{ge^{-\bfrac{E}{k_\mathrm{B}T}}}{\sum_E ge^{-\bfrac{E}{k_\mathrm{B}T}}} \right) \left( \frac{e^{-\bfrac{E_0}{k_\mathrm{B}T}}}{e^{-\bfrac{E_0}{k_\mathrm{B}T}}} \right) = \frac{ge^{-\bfrac{E}{k_\mathrm{B}T}}}{\sum_E ge^{-\bfrac{E}{k_\mathrm{B}T}}} \end{align} $$
Example 2.1
At 298 K, calculate the ratio of the number of \(\chem{NH_3}\) molecules in the excited state to the number in the ground state, where the excited state is (a) the \(0^-\) state of the inversion, which lies \(0.79\,\mathrm{cm}^{-1}\) above the \(0^+\) ground state; and (b) the \(1^+\) state, which lies \(932.43\,\mathrm{cm}^{-1}\) above the \(0^+\). (The wavenumber unit, \(\mathrm{cm}^{-1}\), is conventionally used by spectroscopist as an energy unit, based on the relation between the transition energy in the experiment and the reciprocal wavelength of the photon that induces the transition: \(E_{photon}=\frac{hc}{\lambda}\). Because the energy is inversely proportional to the wavelength, energy is given in units of \(\frac{1}{\text{distance}}\).)
Example 2.2
At a temperature of 1000 K, how many vibrational states of \(\chem{H_2}\) are populated by at least 1% of the molecules, given the vibrational constant \(\omega_e=4395\,\mathrm{cm}^{-1}\) and the vibrational energy (relative to the ground state)of approximately \(E_{vib}=\omega_e\nu\)?
Example 2.3
You discover a molecular system having the energy levels and degeneracies $$ \varepsilon = c\left(n-1\right)^6;\;g=n;\;\text{for }n=1,2,3,\dots\text{ and }k_\mathrm{B}T=400c $$ Evaluate the partition function and calculate \(\mathcal{P}(\varepsilon)\) for each of the four lowest energy levels.
The Ideal Gas Law
The Original Problem - Pressure of an Ideal Gas
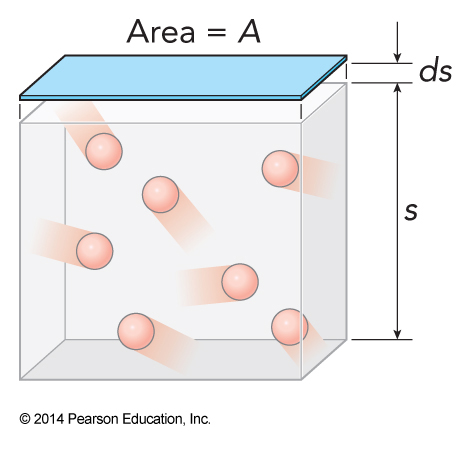
Moving A Wall
- We momentarily free one wall
- It reduces its forces therefore its pressure on the gas, \(P_{min}\)
- The gas pushes the wall out infinitesimally, \(ds\)
- The energy spent moving the wall is \(dE=-F\,dS = -P_{min} A\,dS = -P_{min} \,dV\)
The Pressure Equation
- If we hold \(N\) and \(S\) constant then $$ P_{min} = -\left( \frac{\partial E}{\partial V}\right)_{S,N} $$
- Mostly we will deal with the case where \(P\cong P_{min}\) $$ \text{if}\,P=P_{min}:P=-\left(\frac{\partial E}{\partial V}\right)_{S,N} $$
Cyclic Rule for Partial Derivatives
- The cyclic rule for partial derivatives is $$ \left( \frac{\partial X}{\partial Y}\right)_Z = -\left( \frac{\partial X}{\partial Z}\right)_Y\left(\frac{\partial Z}{\partial Y}\right)_X $$
- This allows us to write $$ P=-\left(\frac{\partial E}{\partial V}\right)_{S,N} = \left(\frac{\partial E}{\partial S}\right)_{V,N} \left(\frac{\partial S}{\partial V}\right)_{E,N} $$
- Another equation for temperature $$ T=\left(\frac{\partial E}{\partial S}\right)_{V,N} $$
Determining Entropy Change with Volume
- Let the translational states of the gas correspond to all the possible ways the gas particles can be arranged inside our box, regardless of what energy the gas has
- The box has a volume \(V\)
- Break up the volume into units \(V_0\), each able to hold a single particle
- We have \(M\) number of units so \(V=MV_0\)
More On Our Gas
- The particles are indistinguishable
- This reduces the number of distinct states
- If we had two particles the number of distinct states would be \(\bfrac{M\left(M-1\right)}{2}\)
- Extending this to \(N\) particles $$ \frac{M(M-1)(M-2)\dots (M-N)}{N!} $$
- For a gas \(M \gg N\)
Even More on Our Gas
- Introduce a constant \(A\) to absorb any other parameter’s effects, such as energy $$ \Omega = \lim_{M\gg N} A\frac{1}{N!} M(M-1)(M_2)\dots (M_N)=A\frac{1}{N!}M^N $$
- Because \(V=MV_0\) $$ \Omega\frac{1}{N!}\left(\frac{V}{V_0}\right)^N = A\frac{1}{V_0^NN!} V^N $$
The Entropy
- Ignoring everything but the volume dependence $$ \begin{align} S &= k_\mathrm{B} \ln \Omega = k_\mathrm{B} \ln \left[ \text{constant}\cdot V^N \right] \\ &= k_\mathrm{B} \left[ \ln \text{constant} + N\ln V\right] \end{align} $$
- This gives $$ \left(\frac{\partial S}{\partial V}\right)_{E,N} = k_\mathrm{B}N\left(\frac{\partial \ln V}{\partial V}\right)_{E,N} = k_\mathrm{B} \frac{N}{V} $$
- So $$ P=\left(\frac{\partial E}{\partial S}\right)_{V,N}\left(\frac{\partial S}{\partial V}\right)_{E,N} = \frac{k_\mathrm{B}TN}{V} $$
So What?
- Remember that \(k_\mathrm{B}\mathcal{N}_\mathrm{A}=R\)
- So $$ \begin{align} P &= \frac{k_\mathrm{B}TN}{V} = \frac{\frac{R}{\mathcal{N}_\mathrm{A}}TN}{V} = \frac{RT\frac{N}{\mathcal{N}_\mathrm{A}}}{V} = \frac{RTn}{V} \\ PV &= nRT \end{align} $$
- We managed to derive the classical ideal gas law equation from statistical mechanics using a canonical ensemble!
/